Executive Summary
Major properties of the lipophilic antiviral dinucleosides:
 Chemistry:
Chemistry:
The novel compounds N4-hexadecyl-2'-deoxyribocytidylyl-(3'-5')-3'-azidothymidine (N4-hexadecyldC-AZT) and N4-hexadecyl-2'-deoxyribo-cytidylyl-(3'-5')-2',3'-
dideoxycytidine (N4-hexadecyldC-ddC) are amphiphilic antiviral drugs which were chemically derived from the inactive lipophilic derivative N4-hexadecyl-2'-deoxyribocytidine and azidothymidine (AZT) or dideoxycytidine (ddC).
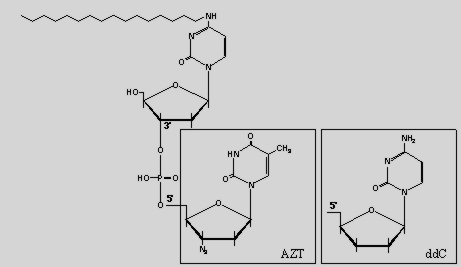 |
Due to the masked phosphate group in these derivatives, the monophosphates of the corresponding antiviral drugs AZT or ddC are released after enzymatic cleavage. As a consequence the cellular pharmacology, pharmacokinetics and mechanisms of action of the dinucleosides are significantly different from those of AZT and ddC respectively.
Chemical synthesis:
The active antiviral molecules AZT and ddC are condensed according to the hydrogenphosphonate method to the lipophilic counterpart N4-hexadecyl-5'-O-(4-monomethoxytrityl)-2'-deoxycytidine-3'-hydrogenphosphonate.
Pharmaceutical formulation:
The dinucleotide derivatives are available as lyophilized preparations of small unilamellar liposomes of 50-100 nm average size. The lyophilized products are of excellent stability. The drugs can be used for parenteral treatment schedules after reconstitution with physiological buffers.
In vivo Activity of N4-hexadecyldC-AZT in the Rauscher leukemia model:
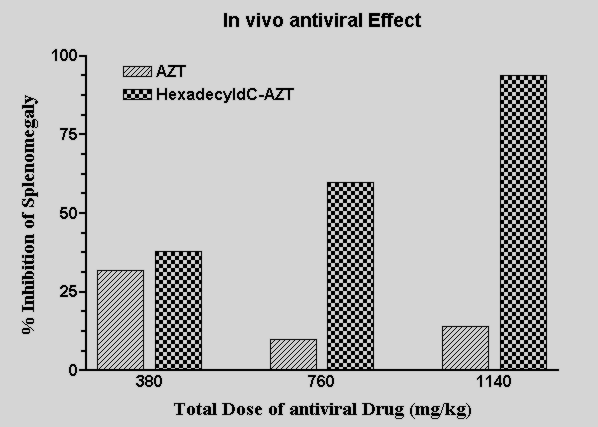 |
Results published in Antiviral Res. 24: 79-93 (1994).
In vitro antiviral effects on HIV-1 infected human lymphocyte cultures:
In HIV-1 infected HeLa cells virus proliferation was inhibited in a similar dose-response manner as AZT. The IC50 values were 0.035 microM for AZT and 0.5 microM for N4-hexadecyldC-AZT. The prodrug nature of the derivative is probably the cause that in vitro higher concentrations than AZT are required to obtain comparable antiviral effects.
Hematological toxicity of N4-hexadecyldC-AZT:
We found that the AZT-derivative has amphiphilic properties and that it has therefore a hemolytic activity. For in vivo applications N4-hexadecyldC-AZT has therefore to be formulated in liposomes which, - as shown with the in vivo experiments, - completely abolishes the unwanted hemolytic activity of the drug.
Preclinical Pharmacokinetics:
Pharmacokinetics of N4-hexadecyldC-AZT in healthy mice gave plasma half-lives of t1/2(a) = 14 min and t1/2(b) = 3.5 hours. The area under the curve (AUC) was 7-fold increased as compared to that of AZT. A relatively high proportion of the drug is found in the liver, however with similar elimination kinetics as in plasma.
Cellular Pharmacology:
Incubations of N4-hexadecyldC-AZT with CEM4 cells showed that in contrast to AZT the drug is also distributed into membrane fractions of the cells, whereas the cytoplasmatic fraction is reduced to about half of that of AZT.
Mechanisms of action:
N4-hexadecyldC-AZT is probably a prodrug of AZT. Due to its amphiphilic properties the drug has pharmacokinetic and biopharmaceutic properties which are significantly different from those of AZT. It is conceivable that with this drug HIV reservoirs in lymphatic organs might be reached. Similar results as shown here for the AZT-derivative were obtained with the ddC-derivative.
Conclusions:
From the presently known pharmacological data it can be concluded that these new AZT- and ddC-dinucleotides might possess a high potential for the treatment of HIV. However, to prove their activity, it is necessary to conduct further experiments.
Literature References:
- Peghini, P.A., Zahner, R., Schott, H., and Schwendener, R.A. Anti-HIV and anti-hepatitis B activities in vitro and pharmacokinetic properties of heterodinucleoside phosphates containing AZT or ddC. Antivir. Chem. & Chemother., 9 : 117-126 (1998).
- Schott, H., Häussler, M.P., Gowland, P., Bender, A.,von Briesen, H., Schwendener, R.A. Synthesis, and antiviral Properties in vitro of amphiphilic Dinucleoside Phosphate Derivatives of 2',3'-Dideoxycyticine (ddC). Antivir. Chem. & Chemother.,6: 320-326 (1995).
- Schott, H., Häussler, M.P., Gowland, P., Horber, D.H., Schwendener, R.A. Synthesis, and some properties of, amphiphilic dinucleoside phosphate derivatives of 3'-azido-2',3'-dideoxy-thymidine (AZT).Antivir. Chem. & Chemother. 5: 387-394 (1994).
- Schwendener, R.A., Gowland, P., Horber, D.H., Kettner, A., Zahner, R., Schertler, A., and Schott, H., New lipophilic alkyl/acyl Dinucleoside Phosphates as Prodrugs of 3'-Azido-2',3'-dideoxythymidine. Inhibition of HIV-1 Replication in vitro and antiviral Effect against Rauscher Leukemia Virus infected Mice with delayed Treatment Regimens.Antiviral Research,24: 79-93 (1994).
- Schott, H., Häussler, M.P., and Schwendener, R.A. Synthese und Eigenschaften von 4-hexadecyl-2'-desoxycytidylyl-(3'-5')-5-ethyl-2'- desoxyuridin und 2'-Desoxy-thymidylyl-(3'-5')-N4-hexadecyl-1-beta-D-arabinofuranosylcytosin, zwei Vertreter einer neuen Prodrug-Gruppe.Liebigs Annalen der Chemie, 277-282 (1994).
- Peghini, P.A., Zahner, R., Schott, H., and Schwendener, R.A. Anti-HIV and anti-hepatitis B activities in vitro and pharmacokinetic properties of heterodinucleoside phosphates containing AZT or ddC. Antivir. Chem. & Chemother., 9 : 117-126 (1998).
- Schott, H., Häussler, M.P., Gowland, P., Bender, A.,von Briesen, H., Schwendener, R.A. Synthesis, and antiviral Properties in vitro of amphiphilic Dinucleoside Phosphate Derivatives of 2',3'-Dideoxycyticine (ddC). Antivir. Chem. & Chemother.,6: 320-326 (1995).
- Schott, H., Häussler, M.P., Gowland, P., Horber, D.H., Schwendener, R.A. Synthesis, and some properties of, amphiphilic dinucleoside phosphate derivatives of 3'-azido-2',3'-dideoxy-thymidine (AZT).Antivir. Chem. & Chemother. 5: 387-394 (1994).
- Schwendener, R.A., Gowland, P., Horber, D.H., Kettner, A., Zahner, R., Schertler, A., and Schott, H., New lipophilic alkyl/acyl Dinucleoside Phosphates as Prodrugs of 3'-Azido-2',3'-dideoxythymidine. Inhibition of HIV-1 Replication in vitro and antiviral Effect against Rauscher Leukemia Virus infected Mice with delayed Treatment Regimens.Antiviral Research,24: 79-93 (1994).
- Schott, H., Häussler, M.P., and Schwendener, R.A. Synthese und Eigenschaften von 4-hexadecyl-2'-desoxycytidylyl-(3'-5')-5-ethyl-2'- desoxyuridin und 2'-Desoxy-thymidylyl-(3'-5')-N4-hexadecyl-1-beta-D-arabinofuranosylcytosin, zwei Vertreter einer neuen Prodrug-Gruppe.Liebigs Annalen der Chemie, 277-282 (1994).
Back to Top
Go to the other pages: