Navigation auf uzh.ch
Navigation auf uzh.ch
One of the best known processes of tumorigenesis in humans is that which occurs in the colon (or large intestine). Thanks to major advances achieved in the last three decades in the fields of endoscopy, histology and molecular pathology, cancer of the large intestine is no longer viewed as a single disease entity: several distinct phenotypes have been identified, and this phenotypic variability is already evident in the precancerous lesions that develop in the gut mucosa. Even today, these lesions are often referred to collectively as colorectal polyps. However, although most of these premalignant lesions are raised, polyp-like growths, more recent research has revealed that there are others that are only slightly elevated above the mucosal surface, flat, or even depressed like a crater. Polyps are much easier to see during routine colonoscopy, and that is one reason they have received so much attention. But the nonpolypoid lesions are now being identified with increasing frequency, in part because clinicians are becoming more aware of their existence and importance, and in part because of the development of more sensitive endoscopic techniques.
Precancerous colorectal lesions are also collectively referred to as adenomas. This term refers to the pattern of cellular dysplasia seen by the pathologist who examines the lesion under a microscope. The adenomatous pattern is very common in precancerous colorectal lesions, but it is not the only pattern. Some benign lesions have cells that are arranged in a saw-toothed or serrated pattern, and they seem to give rise to a particular colorectal cancer phenotype.
The phenotype of a tumor is the outward expression of the specific genetic and epigenetic alterations found in the tumor cells. Some of these somatic alterations have already been well defined; others have been partially characterized, and many have yet to be identified. Changes affecting the genes have wide-ranging effects that are not limited to the appearance of the tumor and the arrangement of its cells: they also determine the tumor behaviour, its aggressiveness and responsiveness to anti-cancer drugs. Thanks to the availability of high-throughput analytical tools (genomics, epigonomics, transcriptomics, proteomics, metabolomics etc.), we can now identify, in each colon tumor tissue, a vast number of molecular characteristics that produce these phenotypic features– and this is an essential step toward individualized (and hopefully more effective) treatment regimens.
Diagn Pathol 16, 4 (2021); https://doi.org/10.1186/s13000-020-01064-1
Giancarlo Marra.
Background: Approximately 60% of colorectal cancer (CRC) precursor lesions are the genuinely-dysplastic conventional adenomas (cADNs). The others include hyperplastic polyps (HPs), sessile serrated lesions (SSL), and traditional serrated adenomas (TSAs), subtypes of a class of lesions collectively referred to as “serrated.” Endoscopic and histologic differentiation between cADNs and serrated lesions, and between serrated lesion subtypes can be difficult. Methods: We used in situ hybridization to verify the expression patterns in CRC precursors of 21 RNA molecules that appear to be promising differentiation markers on the basis of previous RNA sequencing studies. Results: SSLs could be clearly differentiated from cADNs by the expression patterns of 9 of the 12 RNAs tested for this purpose (VSIG1, ANXA10, ACHE, SEMG1, AQP5, LINC00520, ZIC5/2, FOXD1, NKD1). Expression patterns of all 9 in HPs were similar to those in SSLs. Nine putatively HP-specific RNAs were also investigated, but none could be confirmed as such: most (e.g., HOXD13 and HOXB13), proved instead to be markers of the normal mucosa in the distal colon and rectum, where most HPs arise. TSAs displayed mixed staining patterns reflecting the presence of serrated and dysplastic glands in the same lesion. Conclusions: Using a robust in situ hybridization protocol, we identified promising tissue-staining markers that, if validated in larger series of lesions, could facilitate more precise histologic classification of CRC precursors and, consequently, more tailored clinical follow-up of their carriers. Our findings should also fuel functional studies on the pathogenic significance of specific gene expression alterations in the initiation and evolution of CRC precursor subtypes.
Supported by the Swiss National Science Foundation grant to GM (no. 310030_179477 / 1).
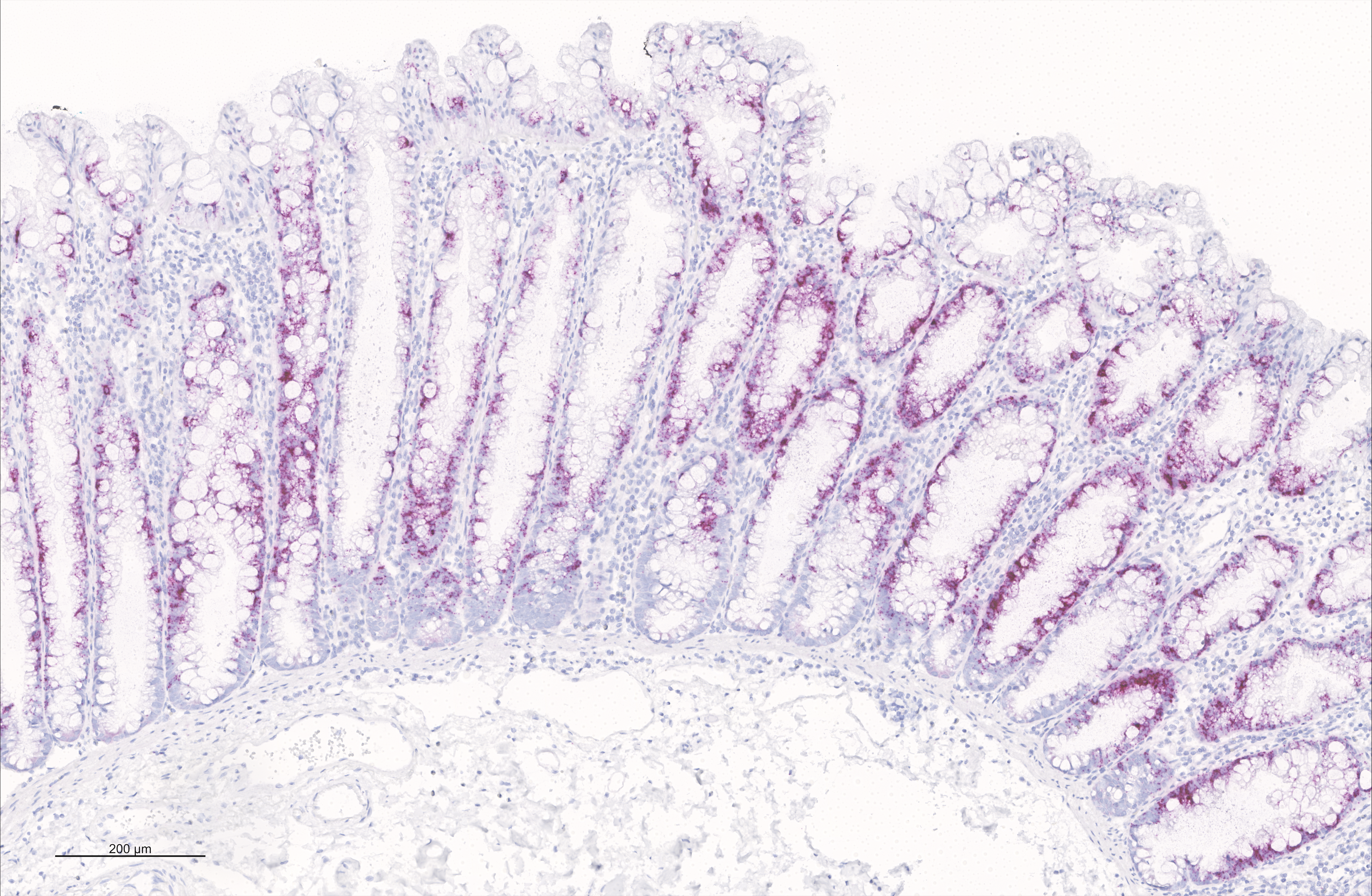
Figure - In situ hybridization analysis of VSIG1 mRNA expression in a sessile serrated lesion of the large intestine.
Epigenetics 2021; https://doi.org/10.1186/s13000-020-01064-1
Physiological aging and tumorigenesis are both associated with epigenomic alterations in human tissue cells, the most extensively investigated of which entails de novo cytosine methylation (i.e., hypermethylation) within the CpG dinucleotides of CpG islands. Genomic regions that become hypermethylated during tumorigenesis are generally believed to overlap regions that acquire methylation in normal tissues as an effect of aging. To define the extension of this overlap, we analyzed the DNA methylomes of 48 large-bowel tissue samples taken from women of different ages during screening colonoscopy: 18 paired samples of normal and lesional tissues from donors harboring a precancerous lesion and 12 samples of normal mucosa from tumor-free donors. Each sample was subjected to targeted, genome-wide bisulfite sequencing of ~2.5% of the genome, including all CpG islands. In terms of both its magnitude and extension along the chromatin, tumor-associated DNA hypermethylation in these regions was much more conspicuous than that observed in the normal mucosal samples from older (vs. younger) tumor-free donors. 83% of the aging-associated hypermethylated regions (n=2501) coincided with hypermethylated regions observed in tumor samples. However, 86% of the regions displaying hypermethylation in precancerous lesions (n=16,772) showed no methylation changes in the aging normal mucosa. The tumor-specificity of this latter hypermethylation was validated using published sets of data on DNA methylation in normal and neoplastic colon tissues. This extensive set of genomic regions displaying tumor-specific hypermethylation represents a rich vein of putative biomarkers for the early, noninvasive detection of colorectal tumors in women of all ages.
Supported by the Swiss National Science Foundation grant no. 310030_179477/1 to S.O., S.S. and G.M., and Novartis Foundation for medical-biological research, grant no. 17B086 to H.R.P.
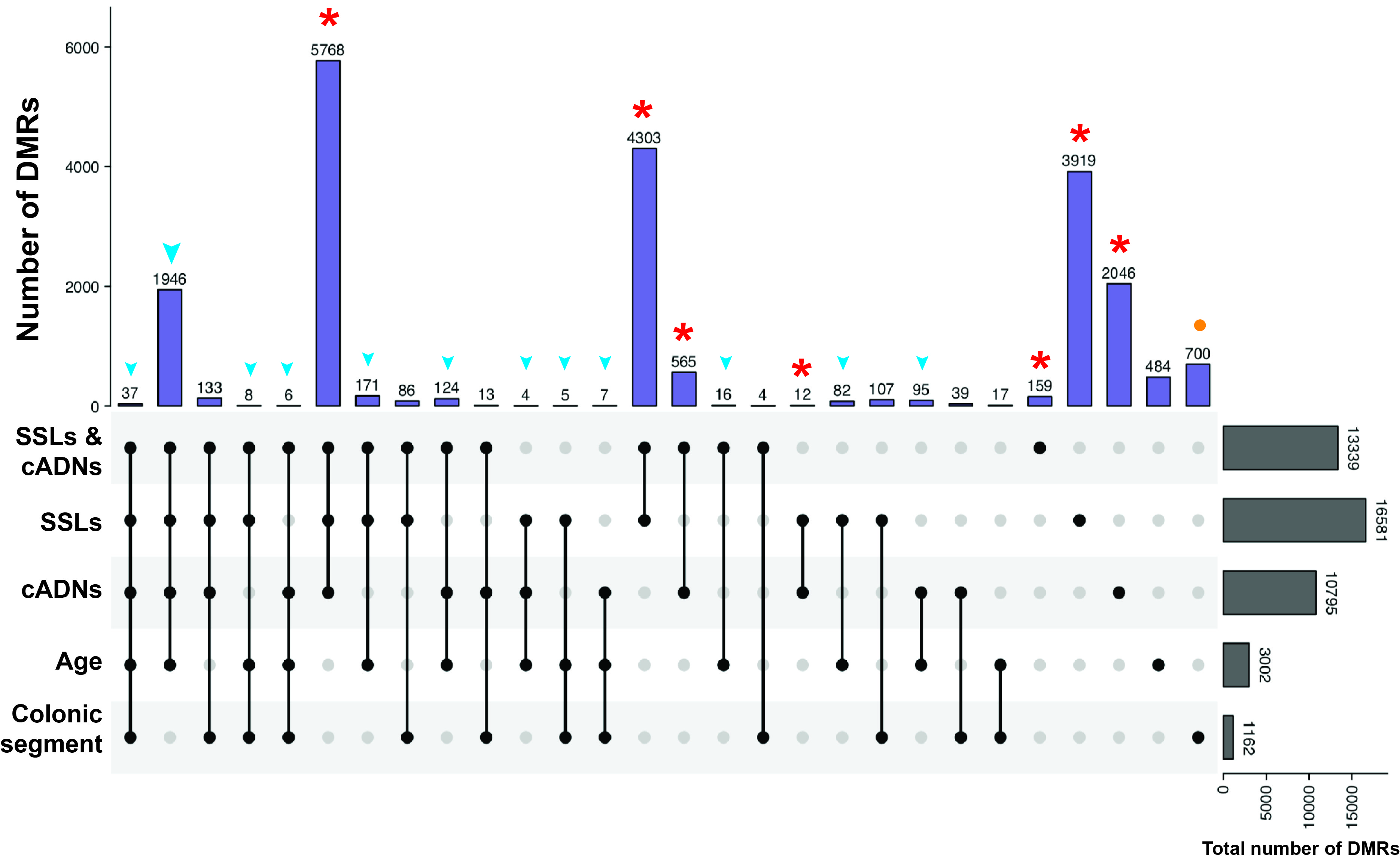
Figure 3B of the paper - UpSet plot showing the tumorigenesis-, aging-, and colon segment-specificities of the hypermethylated DMRs. The vast majority of the hypermethylated DMRs in tumors (in SSLs, cADNs, or both, red asterisks) were tumorigenesis-specific, i.e., regions that displayed no aging-related or colon segment-related changes in the NM. In contrast, over 80% of the aging-associated DMRs overlapped tumorigenesis-associated DMRs (blue arrowheads). Most regions that were differentially methylated in SIG vs. CEC NM samples displayed no methylation alterations associated with aging or tumorigenesis (orange dots). (Abbreviations as defined in Figure 1.)
DAMEfinder: a method to detect differential allele-specific methylation
Epigenetics & Chromatin (2020) 13:25; https://doi.org/10.1186/s13072-020-00346-8
Stephany Orjuela, Dania Machlab, Mirco Menigatti, Giancarlo Marra and Mark D. Robinson.
Background: DNA methylation is a highly studied epigenetic signature that is associated with regulation of gene expression, whereby genes with high levels of promoter methylation are generally repressed. Genomic imprinting occurs when one of the parental alleles is methylated, i.e., when there is inherited allele-specific methylation (ASM). A special case of imprinting occurs during X chromosome inactivation in females, where one of the two X chromosomes is silenced, to achieve dosage compensation between the sexes. Another more widespread form of ASM is sequence dependent (SD-ASM), where ASM is linked to a nearby heterozygous single nucleotide polymorphism (SNP). Results: We developed a method to screen for genomic regions that exhibit loss or gain of ASM in samples from two conditions (treatments, diseases, etc.). The method relies on the availability of bisulfite sequencing data from multiple samples of the two conditions. We leverage other established computational methods to screen for these regions within a new R package called DAMEfinder. It calculates an ASM score for all CpG sites or pairs in the genome of each sample, and then quantifies the change in ASM between conditions. It then clusters nearby CpG sites with consistent change into regions. In the absence of SNP information, our method relies only on reads to quantify ASM. This novel ASM score compares favorably to current methods that also screen for ASM. Not only does it easily discern between imprinted and non-imprinted regions, but also females from males based on X chromosome inactivation. We also applied DAMEfinder to a colorectal cancer dataset and observed that colorectal cancer subtypes are distinguishable according to their ASM signature. We also re-discover known cases of loss of imprinting. Conclusion: We have designed DAMEfinder to detect regions of differential ASM (DAMEs), which is a more refined definition of differential methylation, and can therefore help in breaking down the complexity of DNA methylation and its influence in development and disease.
Supported by the Swiss National Science Foundation grant no. 310030-160163/1 to S.O. and G.M., and the University Research Priority Program Evolution in Action at the University of Zurich to M.D.R.
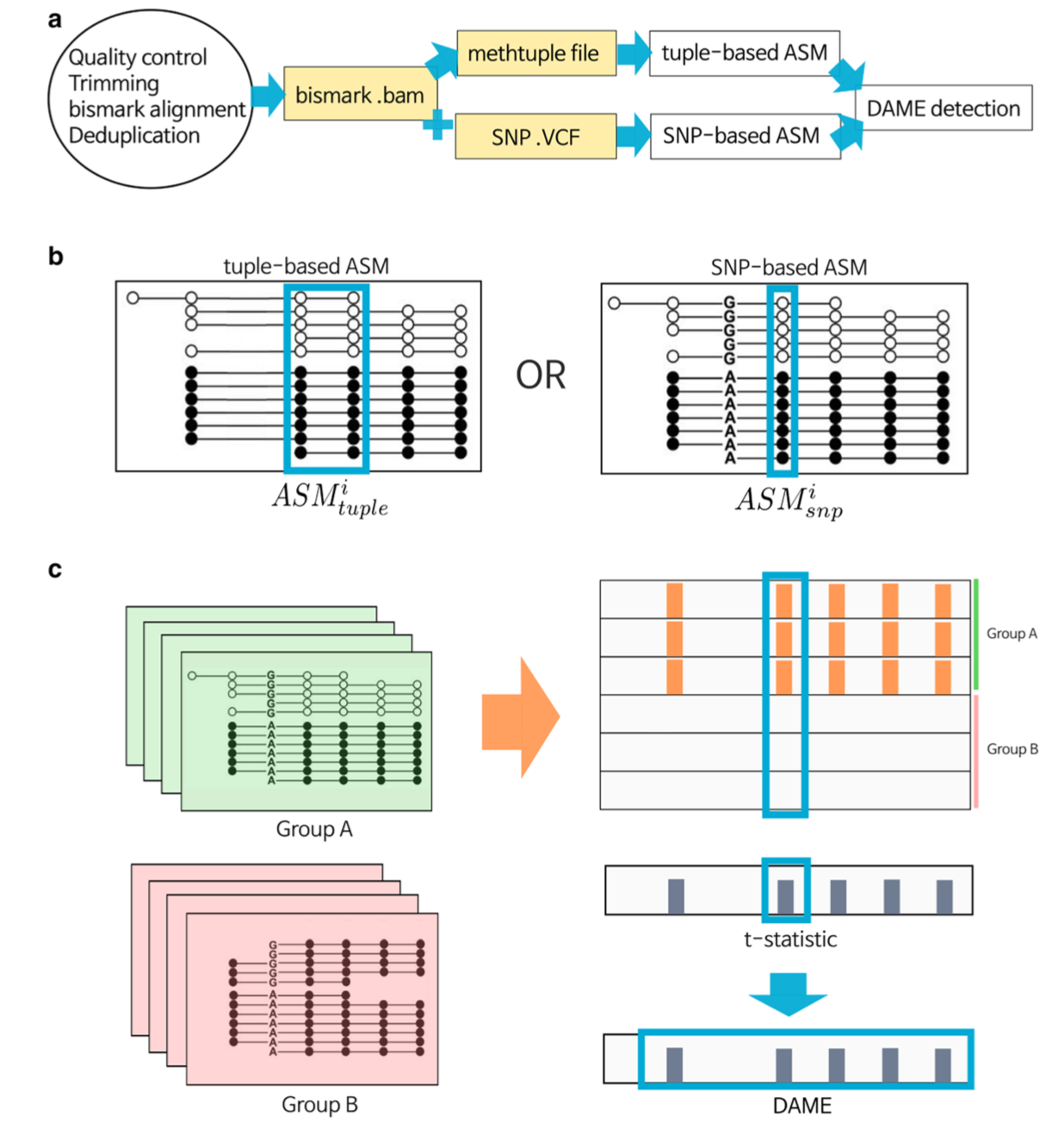
Figure 1 of the paper - The DAMEfinder pipeline. a) Files necessary to run DAMEfinder are reported in yellow rectangles. White rectangles show the main R outputs from DAMEfinder. Steps to be run before DAMEfinder are in the circle, i.e., fastq files undergo quality control and read alignment with bismark (ref. 43). The resulting bam file is used to calculate an ASM score, which can be done in two ways: b) (i) the tuple-based strategy that takes as input a beforehand created methtuple (ref. 41) file. The score is calculated based on the read counts of pairs of CpG sites. (ii) the SNP-based strategy, which takes as input both the bam file and a VCF file with heterozygous SNPs. Here, the score is calculated for each CpG site in the reads containing a SNP. c) We determine differential ASM by calculating a statistic based on either the tuple ASM or the SNP-ASM (using limma [ref. 39]), which reflects the difference between two conditions (Group A vs. Group B) for each genomic position (tuple or site). DAMEs are defined based on this statistic, as regions of contiguous positions with a consistent change in ASM.
The DNA hypermethylation phenotype of colorectal cancer liver metastases resembles that of the primary colorectal cancers.
BMC Cancer (2020) 20:290; https://doi.org/10.1186/s12885-020-06777-6
Stephany Orjuela, Mirco Menigatti, Peter Schraml, Patryk Kambakamba, Mark D. Robinson, and Giancarlo Marra.
Background: Identifying molecular differences between primary and metastatic colorectal cancers—now possible with the aid of omics technologies—can improve our understanding of the biological mechanisms of cancer progression and facilitate the discovery of novel treatments for late-stage cancer. We compared the DNA methylomes of primary colorectal cancers (CRCs) and CRC metastases to the liver. Laser microdissection was used to obtain epithelial tissue (10 to 25 x 106 µm2) from sections of fresh-frozen samples of primary CRCs (n=6), CRC liver metastases (n=12), and normal colon mucosa (n=3). DNA extracted from tissues was enriched for methylated sequences with a methylCpG binding domain (MBD) polypeptide-based protocol and subjected to deep sequencing. The performance of this protocol was compared with that of targeted enrichment for bisulfite sequencing used in a previous study of ours.
Results: MBD enrichment captured a total of 322,551 genomic regions (249.5 Mb or ~7.8% of the human genome), which included over seven million CpG sites. A few of these regions were differentially methylated at an expected false discovery rate (FDR) of 5% in neoplastic tissues (primaries: 0.67%, i.e., 2155 regions containing 279,441 CpG sites; liver metastases: 1%, i.e., 3223 regions containing 312,723 CpG sites) as compared with normal mucosa samples. Most of the differentially methylated regions (DMRs; 94% in primaries; 70% in metastases) were hypermethylated, and almost 80% of these (1882 of 2396) were present in both lesion types. At 5% FDR, no DMRs were detected in liver metastases vs. primary CRC. However, short regions of low-magnitude hypomethylation were frequent in metastases but rare in primaries. Hypermethylated DMRs were far more abundant in sequences classified as intragenic, gene-regulatory, or CpG shelves-shores-island segments, whereas hypomethylated DMRs were equally represented in extragenic (mainly, open-sea) and intragenic (mainly, gene bodies) sequences of the genome. Compared with targeted enrichment, MBD capture provided a better picture of the extension of CRC-associated DNA hypermethylation but was less powerful for identifying hypomethylation.
Conclusions: Our findings demonstrate that the hypermethylation phenotype in CRC liver metastases remains similar to that of the primary tumor, whereas CRC-associated DNA hypomethylation probably undergoes further progression after the cancer cells have migrated to the liver.
Supported by the Swiss National Science Foundation grant no. 310030-179477 to S.O. and G.M.; the Swiss Cancer League grant no. 3397-02-2014 to M.M., and the University Research Priority Program Evolution in Action at the University of Zurich to M.D.R.
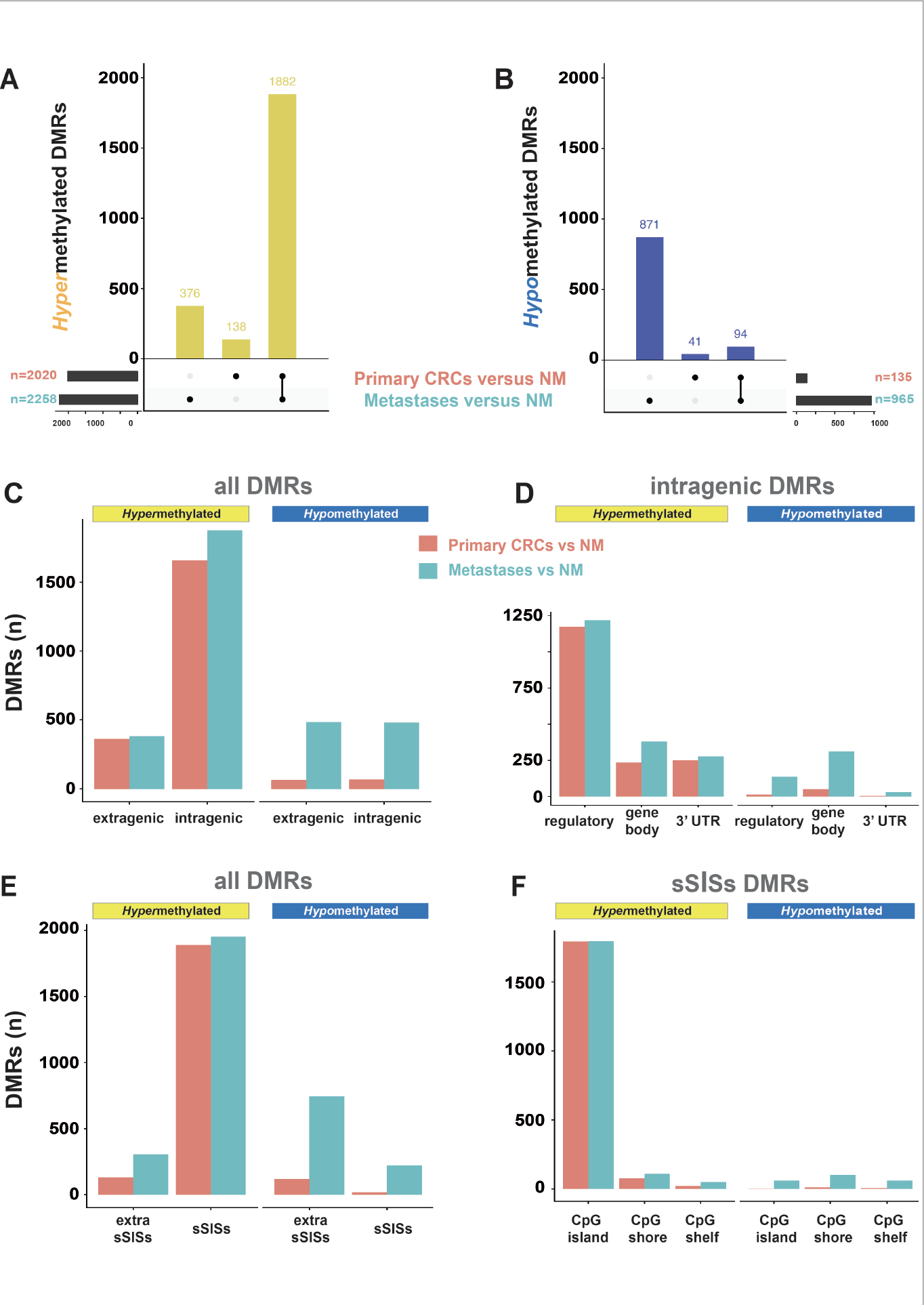
Figure 2 of the paper. Genomic regions in primary CRCs or CRC liver metastases displaying differential methylation relative to that in normal colon mucosa (NM). A and B. Differentially methylated regions (DMRs) characterized by hypermethylation and hypomethylation (vs. NM) present in primary CRCs, metastatic CRCs, or both. C, D, E, and F. Distributions of hypermethylated and hypomethylated DMRs in the extragenic vs. intragenic genomes; among the intragenic genome components; between the sSISs (i.e., CpG island and flanking CpG shores and shelves) and extra-sSISs genomic segments, and among the sSISs components, respectively. (See Figure 1 of the paper for topography of genomic segments.)
Hannah R. Parker, Stephany Orjuela, Andreia Martinho Oliveira, Fabrizio Cereatti, Matthias Sauter, Henriette Heinrich, Giulia Tanzi, Achim Weber, Paul Komminoth, Stephan Vavricka, Luca Albanese, Federico Buffoli, Mark D. Robinson, and Giancarlo Marra.
Sessile serrated adenomas/polyps (SSA/Ps) are the putative precursors of the ~20% of colon cancers with the CpG island methylator phenotype (CIMP), but their molecular features are poorly understood. We used high-throughput analysis of DNA methylation and gene expression to investigate the epigenetic phenotype of SSA/Ps. Fresh-tissue samples of 17 SSA/Ps and (for comparison purposes) 15 conventional adenomas (cADNs)—each with a matched sample of normal mucosa—were prospectively collected during colonoscopy (total no. samples analyzed: 64). DNA and RNA were extracted from each sample. DNA was subjected to bisulfite next-generation sequencing to assess methylation levels at ~2.7 million CpG sites located predominantly in gene regulatory regions and spanning 80.5Mb (~2.5% of the genome); RNA was sequenced to define the samples’ transcriptomes. An independent series of 61 archival lesions was used for targeted verification of DNA methylation findings. Compared with normal mucosa samples, SSA/Ps and cADNs exhibited markedly remodeled methylomes. In cADNs, hypomethylated regions were far more numerous (18,417 vs 4288 in SSA/Ps) and rarely affected CpG islands/shores. SSA/Ps seemed to have escaped this wave of demethylation. Cytosine hypermethylation in SSA/Ps was more pervasive (hypermethylated regions: 22,147 vs 15,965 in cADNs; hypermethylated genes: 4938 vs 3443 in cADNs) and more extensive (region for region), and it occurred mainly within CpG islands and shores. Given its resemblance to the CIMP typical of SSA/Ps' putative descendant colon cancers, we refer to the SSA/P methylation phenotype as proto-CIMP. Verification studies of six hypermethylated regions (3 SSA/P-specific and 3 common) demonstrated the high potential of DNA methylation markers for predicting the diagnosis of SSA/Ps and cADNs. Surprisingly, proto-CIMP in SSA/Ps was associated with upregulated gene expression (n=618 genes vs 349 that were downregulated); downregulation was more common in cADNs (n=712 vs 516 upregulated genes). In conclusion, the epigenetic landscape of SSA/Ps differs markedly from that of cADNs. These differences are a potentially rich source of novel tissue-based and noninvasive biomarkers that can add precision to the clinical management of the two most frequent colon-cancer precursors.
Supported by grants of the Swiss National Science Foundation (grant no. 310030-160163/1) and of the Novartis Foundation for medical-biological Research.
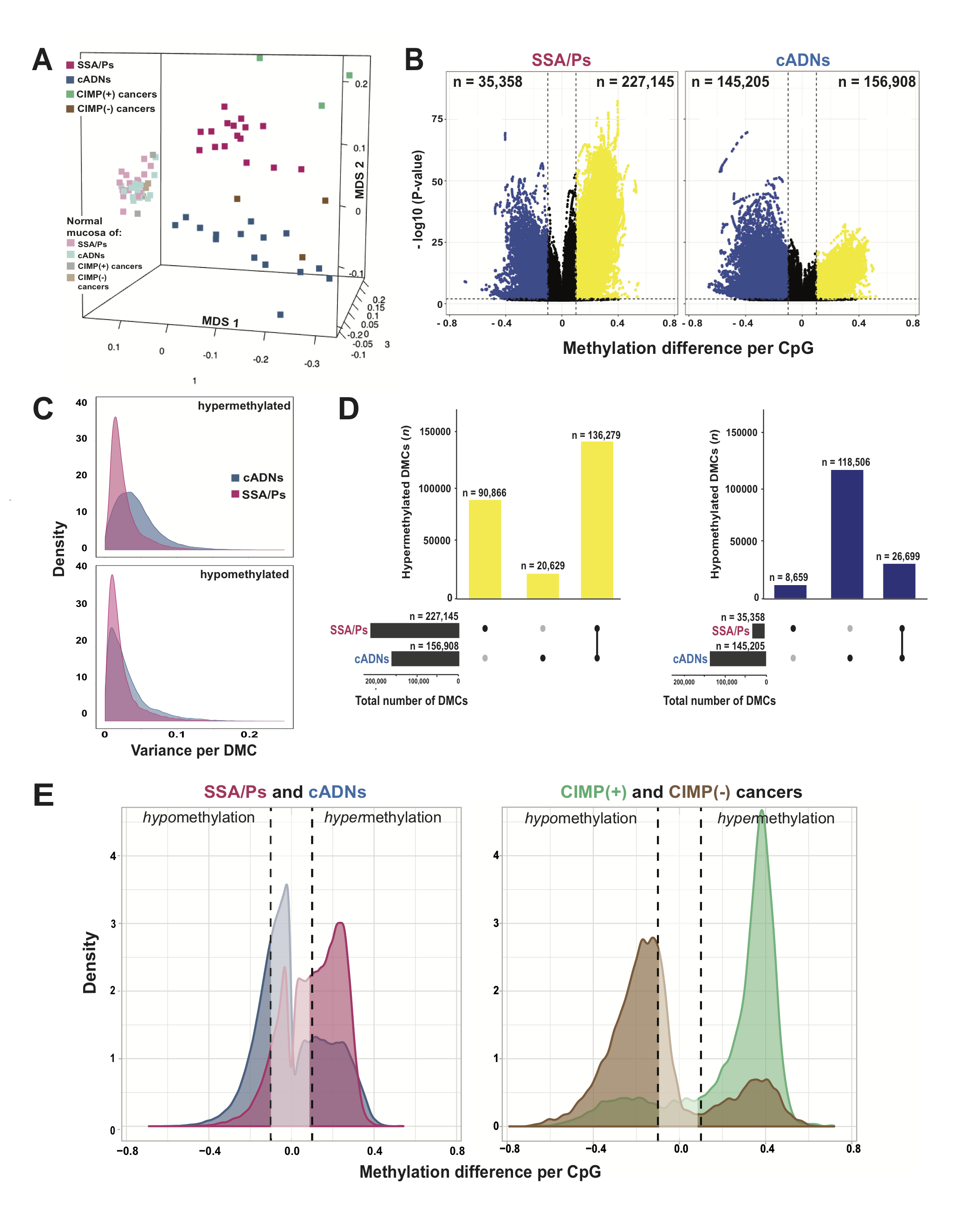
Figure 1 of the paper: Differentially methylated cytosines (DMCs) in precancerous and cancerous colon lesions. A. MDS plot of DNA methylation levels in SSA/Ps (n=16); cADNs (n=15); CIMP(+) cancers (n=3); CIMP(-) cancers (n=3); and matched samples of normal mucosa for each lesion. B. Volcano plots showing the magnitude (x axis) and statistical significance (y axis) of the differential methylation observed at DMCs identified in SSA/Ps and cADNs. X-axis: The magnitude of differential methylation was calculated as the M:T ratio (no. methylated reads / total no. reads) for the tumor sample minus M:T ratio for matched normal-tissue control. Black dots: DMCs with absolute methylation differences of <0.1 and P-values >0.01. Yellow and blue dots: highly significant (P-value <0.01) DMCs (hypermethylated and hypomethylated, respectively). C. Density plot showing variance at the hypermethylated (top) and hypomethylated (bottom) DMCs (yellow and blue dots of panel B, respectively). D. UpSet plots showing hypermethylated (left) and hypomethylated (right) DMC sets in SSA/Ps and cADNs and their overlaps. Exact numbers of lesion-specific (•) and shared (•-•) DMCs appear above the bars. E. Overlaid density plots showing the distributions of hypo- and hypermethylated DMCs in SSA/Ps and cADNs (left), and CIMP(+) and CIMP(-) cancers (right). X-axis as described in panel B.
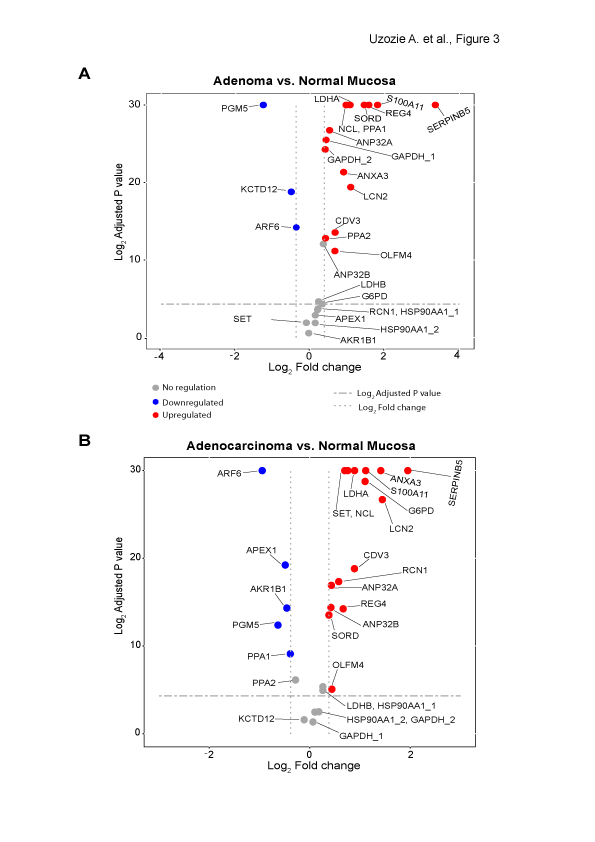
Figure 3 of the paper: Quantification of tumor markers in colorectal adenomas (A) and adenocarcinomas (B) (relative to matched normal mucosal samples). Proteins with an adjusted p value < 0.05 and a fold change ≥ ±1.3 were considered significant. Housekeeping proteins monitored in the two time-scheduled methods are denoted with _1 and _2.
Mol Cell Proteomics. 2014 May;13(5):1198-218
Uzozie A, Nanni P, Staiano T, Grossmann J, Barkow-Oesterreicher S, Shay JW, Tiwari A, Buffoli F, Laczko E, Marra G.
Colorectal adenomas are cancer precursor lesions of the large bowel. A multitude of genomic and epigenomic changes have been documented in these preinvasive lesions, but their impact on the protein effectors of biological function has not been comprehensively explored. Using shotgun quantitative MS, we exhaustively investigated the proteome of 30 colorectal adenomas and paired samples of normal mucosa. Total protein extracts were prepared from these tissues (prospectively collected during colonoscopy) and from normal (HCEC) and cancerous (SW480, SW620, CACO2, HT29, CX1) colon epithelial cell lines. Peptides were labeled with isobaric tags (iTRAQ 8-plex), separated by OFFGEL electrophoresis, and analyzed by LC-coupled tandem MS. Non-redundant protein families (4325 in tissues, 2017 in cell lines) were identified and quantified. Principal component analysis of the results clearly distinguished adenomas from normal mucosal samples, and cancer cell lines from HCEC cells. Two hundred twelve proteins displayed significant adenoma-related expression changes (q-value < 0.02, mean fold change vs. normal mucosa +/-1.4), which correlated (r=0.74) with similar changes previously identified by our group at the transcriptome level. Fifty-one (~25%) proteins displayed directionally similar expression changes in colorectal cancer cells (vs. HCEC cells) and were therefore attributed to the epithelial component of adenomas. Although benign, adenomas already exhibited cancer-associated proteomic changes: 69 (91%) of the 76 protein upregulations identified in these lesions have already been reported in cancers. One of the most striking changes involved sorbitol dehydrogenase (SORD), a key enzyme in the polyol pathway. Validation studies revealed dramatically increased SORD concentrations and activity in adenomas and cancer cell lines, along with important changes in the expression of other enzymes in the same (AKR1B1) and related (KHK) pathways. Dysregulated polyol metabolism may represent a novel facet of the metabolome remodeling associated with tumorigenesis.
Supported by a grant from the Swiss National Science Foundation to G. Marra
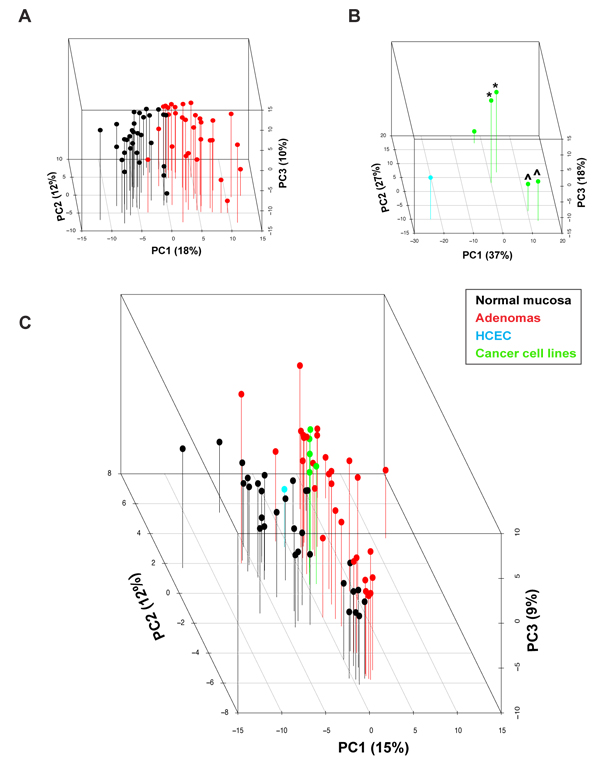
Figure 3 of the paper: Three-dimensional principal component analysis score plot of log2 protein expression intensity values for (A) tissues (normal mucosa, black; adenomas, red), (B) cell lines (HCEC, cyan; colon cancer cell lines, green), and (C) both.
BMC Cancer. 2014 Jan 29;14:46
Vonlanthen J, Okoniewski MJ, Menigatti M, Cattaneo E, Pellegrini-Ochsner D, Haider R, Jiricny J, Staiano T, Buffoli F, Marra G
Background: Biological processes are controlled by transcription networks. Expression changes of transcription factor (TF) genes in precancerous lesions are therefore crucial events in tumorigenesis. Our aim was to obtain a comprehensive picture of these changes in colorectal adenomas.
Methods: Using a 3-pronged selection procedure, we analyzed transcriptomic data on 34 human tissue samples (17 adenomas and paired samples of normal mucosa, all collected with ethics committee approval and written, informed patient consent) to identify TFs with highly significant tumor-associated gene expression changes whose potential roles in colorectal tumorigenesis have been under-researched. Microarray data were subjected to stringent statistical analysis of TF expression in tumor vs. normal tissues, MetaCore-mediated identification of TF networks displaying enrichment for genes that were differentially expressed in tumors, and a novel quantitative analysis of the publications examining the TF genes’ roles in colorectal tumorigenesis.
Results: The 261 TF genes identified with this procedure included DACH1, which plays essential roles in the proper proliferation and differentiation of retinal and leg precursor cell populations in Drosophila melanogaster. Itspossible roles in colorectal tumorigenesis are completely unknown, but it was found to be markedly overexpressed (mRNA and protein) in all colorectal adenomas and in most colorectal carcinomas. However, DACH1 expression was absent in some carcinomas, most of which were DNA mismatch-repair deficient. When networks were built using the set of TF genes identified by all three selection procedures, as well as the entire set of transcriptomic changes in adenomas, five hub genes (TGFB1, BIRC5, MYB, NR3C1, and TERT) where identified as putatively crucial components of the adenomatous transformation process.
Conclusion: The transcription-regulating network of colorectal adenomas (compared with that of normal colorectal mucosa) is characterized by significantly altered expression of over 250 TF genes, many of which have never been investigated in relation to colorectal tumorigenesis.
Keywords: Transcription factors; Gene expression; Colorectal adenomas; DACH1.
Supported by a grant from the Swiss National Science Foundation to G. Marra
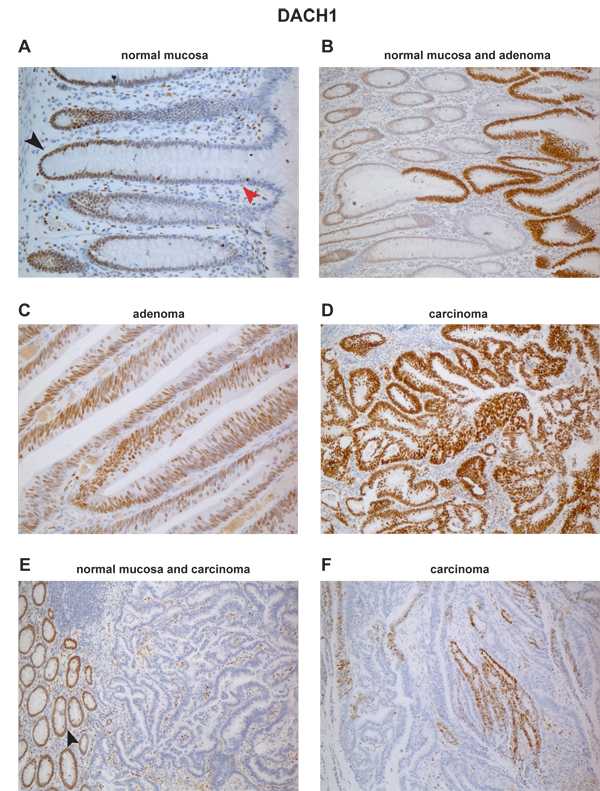
Figure 5 of the paper: Immunohistochemical staining for DACH1 protein in normal and neoplastic colon. (A) In normal mucosa, DACH1 expression is present in the nuclei of proliferating cells in the lower portion of the epithelial crypts (black arrowhead) and completely absent in the differentiated cells in the upper crypts (red arrowhead). (B) High-level DACH1 expression is seen in rapidly proliferating cells of adenomatous glands taking over normal crypts. Abundant expression is also seen in most cells of a colorectal adenoma (C) and a colorectal carcinoma (D). In another colorectal cancer (E), DACH1 expression is absent in neoplastic glands, although proliferating cells in the normal mucosa and in the tumoral stroma are positive. (F) A third colorectal cancer with patchy staining for DACH1.
PLoS One. 2013 Jul 23;8(7):e69473
Tiwari A, Schneider M, Fiorin A, Haider R, Okoniewski M J, Roschitzki B, Uzozie A, Menigatti M, Jiricny J, Marra G
We previously reported that the expression of KIAA1199 in human colorectal tumors (benign and malignant) is markedly higher than that in the normal colonic mucosa. In this study, we investigated the functions of the protein encoded by this gene, which are thus far unknown. Immunostaining studies were used to reveal its subcellular localization, and proteomic and gene expression experiments were conducted to identify proteins that might interact with KIAA1199 and molecular pathways in which it might play roles. Using colon cancer cell lines, we showed that both endogenous and ectopically expressed KIAA1199 is secreted into the extracellular environment. In the cells, it was found mainly in the perinuclear space (probably the ER) and cell membrane. Both cellular compartments were also over-represented in lists of proteins identified by mass spectrometry as putative KIAA1199 interactors and/or proteins encoded by genes whose transcription was significantly changed by KIAA1199 expression. These proteomic and transcriptomic datasets concordantly link KIAA1199 to several genes/proteins and molecular pathways, including ER processes like protein binding, transport, and folding; and Ca2+, G-protein, ephrin, and Wnt signaling. Immunoprecipitation experiments confirmed KIAA1199’s interaction with the cell-membrane receptor ephrin A2 and with the ER receptor ITPR3, a key player in Ca2+ signaling. By modulating Ca2+ signaling, KIAA1199 could affect different branches of the Wnt network. Our findings suggest it may negatively regulate the Wnt / CTNNB1 signaling, and its expression is associated with decreased cell proliferation and invasiveness.
Supported by grants from the Zurich Cancer League (to G. Marra) and the University Research Priority Program, University of Zurich (to A. Tiwari)
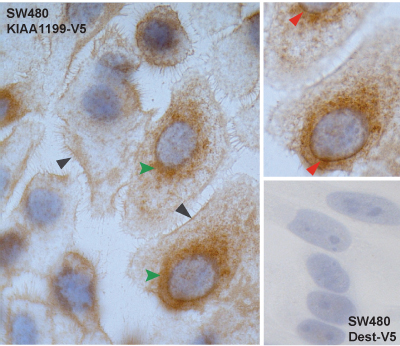
Figure 1 (panel E) of the paper: Immunocytochemical staining of SW480 KIAA1199-V5 colon cancer cells shows KIAA1199 in the cell membrane (gray arrowheads) and perinuclear space (green arrowheads). Upper right inset: Fine focusing clearly reveals staining of the nuclear membrane (red arrowheads). Lower right inset: Negative control (SW480 Dest-V5 immunostained with V5-tag-specific antibody).
BMC Cancer. 2012 Dec 19;12:608
Maglietta R, Liuzzi VC, Cattaneo E, Laczko E, Piepoli A, Panza A, Carella M, Palumbo O, Staiano T, Buffoli F, Andriulli A, Marra G, Ancona N.
Background: The malignant transformation of precancerous colorectal lesions involves progressive alterations at both the molecular and morphologic levels, the latter consisting of increases in size and in the degree of cellular atypia. Analyzing preinvasive tumors of different sizes can therefore shed light on the sequence of these alterations.
Methods: We used a molecular pathway-based approach to analyze transcriptomic profiles of 59 colorectal tumors representing early and late preinvasive stages and the invasive stage of tumorigenesis. A Random Set method was used to identify biological pathways enriched for genes differentially regulated in tumors (compared with 59 samples of normal mucosa).
Results: Of the 880 canonical pathways we investigated, 112 displayed significant tumor-related upregulation or downregulation at one or more stages of tumorigenesis. This allowed us to distinguish between pathways whose dysregulation is probably necessary throughout tumorigenesis and those whose involvement specifically drives progression from one stage to the next. We were also able to pinpoint specific changes within each gene set that seem to play key roles at each transition. The early preinvasive stage was characterized by cell-cycle checkpoint activation triggered by DNA replication stress and dramatic downregulation of basic transmembrane signaling processes that maintain epithelial/stromal homeostasis in the normal mucosa. In late preinvasive lesions, there was also downregulation of signal transduction pathways (e.g., those mediated by G proteins and nuclear hormone receptors) involved in cell differentiation and upregulation of pathways governing nuclear envelope dynamics and the G2>M transition in the cell cycle. The main features of the invasive stage were activation of the G1>S transition in the cell cycle, upregulated expression of tumor-promoting microenvironmental factors, and profound dysregulation of metabolic pathways (e.g., increased aerobic glycolysis, downregulation of pathways that metabolize drugs and xenobiotics). These data provide a launchpad for further exploration of the molecular characterization of colorectal tumorigenesis using systems biology approaches.
Conclusions: Our analysis revealed specific pathways whose dysregulation might play a role in each transition of the transformation process. This is the first study in which such an approach has been used to gain further insights into colorectal tumorigenesis. Therefore, these data provide a launchpad for further exploration of the molecular characterization of colorectal tumorigenesis using systems biology approaches.
Keywords: Colorectal adenoma, Colorectal cancer, Transcriptomics, Molecular pathways, Cell cycle pathways, Random set method
Supported in part by a grant from the Swiss National Science Foundation to G. Marra
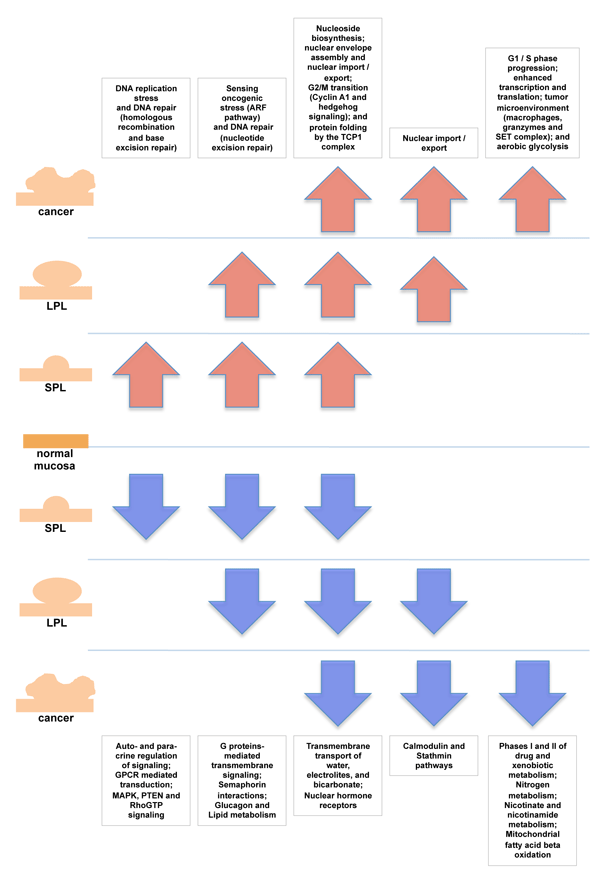
Figure 4 of the paper: Overview of tumor-related pathway dysregulation at different stages of transformation. Pathways displaying identical configurations of dysregulation (e.g., upregulated in SPLs and LPLs but not CRCs) have been combined into 10 more general biological groups (white boxes). Arrows indicate type (up vs. down) of dysregulation.
Fam Cancer. 2011 Sep;10(3):605-16
Kovac M, Laczko E, Haider R, Jiricny J, Mueller H, Heinimann K, Marra G.
Deleterious germ-line variants involving the DNA mismatch repair (MMR) genes have been identified as the cause of the hereditary nonpolyposis colorectal cancer syndrome known as the Lynch syndrome, but in numerous familial clusters of colon cancer the cause remains obscure. We analyzed data for 235 German-speaking Swiss families with nonpolyposis forms of colorectal cancer (one of the largest and most ethnically homogeneous cohorts of its kind) to identify the phenotypic features of forms that cannot be explained by MMR deficiency. Based on the results of microsatellite instability analysis and immunostaining of proband tumor samples, the kindreds were classified as MMR-proficient (n = 134, 57%) or MMR-deficient (n = 101, 43%). In 81 of the latter kindreds, deleterious germ-line MMR-gene variants have already been found (62 different variants, including 13 that have not been previously reported), confirming the diagnosis of Lynch syndrome. Compared with MMR-deficient kindreds, the 134 who were MMR proficient were less likely to meet the Amsterdam Criteria II regarding autosomal dominant transmission. They also had primary cancers with later onset and colon-segment distribution patterns resembling those of sporadic colorectal cancers, and they had lower frequencies of metachronous colorectal cancers and extracolonic cancers in general. Although the predisposition to colorectal cancer in these kindreds is probably etiologically heterogeneous, we were unable to identify distinct phenotypic subgroups solely on the basis of the clinical data collected in this study. Further insight, however, is expected to emerge from the molecular characterization of their tumors.
Supported in part by a grant from the Swiss National Science Foundation to G. Marra

Figure 1 of the paper: The diagnostic flow chart used to classify the 235 probands’ families having MMR-deficient or MMR-proficient familial colorectal cancer.
EMBO Mol Med. 2011 Jun;3(6):334-47
Cattaneo E, Laczko E, Buffoli F, Zorzi F, Bianco MA, Menigatti M, Bartosova Z, Haider R, Helmchen B, Sabates-Bellver J, Tiwari A, Jiricny J, Marra G
Improved colonoscopy is revealing precancerous lesions that were frequently missed in the past, and ∼30% of those detected today have nonpolypoid morphologies ranging from slightly raised to depressed. To characterize these lesions molecularly, we assessed transcription of 23,768 genes in 42 precancerous lesions (25 slightly elevated nonpolypoid and 17 pedunculated polypoid), each with corresponding samples of normal mucosa. Nonpolypoid versus polypoid morphology explained most gene expression variance among samples; histology, size, and degree of dysplasia were also linked to specific patterns. Expression changes in polypoid lesions frequently affected cell-cycling pathways, whereas cell-survival dysregulation predominated in nonpolypoid lesions. The latter also displayed fewer and less dramatic expression changes than polypoid lesions. Paradigmatic of this trend was progressive loss through the normal > nonpolypoid > polypoid > cancer sequence of TMIGD1 mRNA and protein. This finding, along with TMIGD1 protein expression patterns in tissues and cell lines, suggests that TMIGD1 might be associated with intestinal-cell differentiation. We conclude that molecular dysregulation in slightly elevated, nonpolypoid, precancerous colorectal lesions may be somewhat less severe than that observed in classic adenomatous polyps.
Supported by a grant from the Swiss National Science Foundation to G. Marra
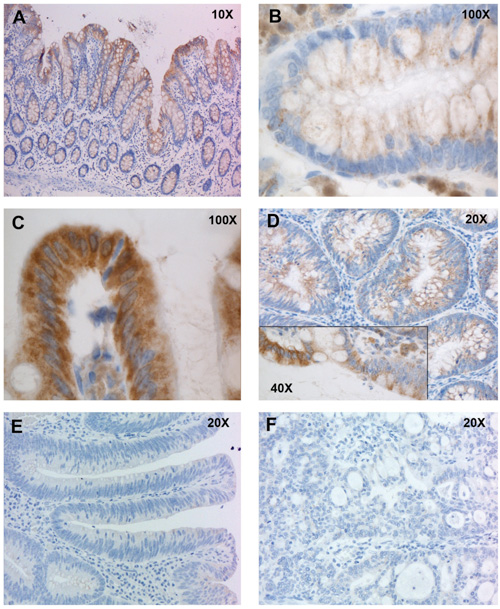
Figure 6 of the paper. Immunohistochemical staining of normal and neoplastic colonic tissues with antibodies against TMIGD1. A. In normal mucosa, TMIGD1 expression is limited to the upper portion of the epithelial crypts, where differentiated cells are located. B. Higher magnification views of TMIGD1 staining at base of a colonic crypt. C. Higher magnification views of TMIGD1 staining at mouth of a colonic crypt. TMIGD1 is located in the cytoplasm and probably in the cell membrane. D. Its expression was markedly reduced in nonpolypoid lesions. The inset shows different levels of expression at the interface between normal (left) and dysplastic (right) epithelium. E. More marked reduction was observed in polypoid lesions, and F. expression was lost in colorectal cancers.